

Inert C-H Bond Transformations Enabled by Organometallic Manganese Catalysis
Yuanyuan Hu, Bingwei Zhou and Congyang Wang*
Traditional organic synthesis relies heavily on the transformations of various preinstalled functional groups, such as cross-coupling reactions using organohalides and organometallic reagents. The strategy of C–H activation enables the direct formation of C–C/C–X (X = heteroatom) bonds from inert C–H bonds, which can enhance the atom- and step-economy of organic synthesis. To date, precious metals have overwhelmingly dominated the C–H activation field; however, the rarity and high cost of these metals necessitate the development of more sustainable catalysts. In this regard, catalysts based on manganese are highly desirable owing to the abundant reserve of manganese in the earth’s crust and its economic benefits, low toxicity, and potentially unique reactivity. Although the first stoichiometric manganese-mediated C–H activation reaction was reported as early as 1970, manganese-catalyzed C–H activation reactions are largely underdeveloped. How to construct an efficient catalytic cycle for manganese in C–H activation reactions remains as a key issue to be addressed.
In this Account, we summarize our recent advances in the manganese-catalyzed transformations of inert C–H bonds. To overcome the challenges associated with building manganese-based catalytic cycles, we developed two novel strategies, namely, synergy between manganese catalysts and bases and between manganese catalysts (with or w/o bases) and acids. By implementing the former strategy, we developed cooperative manganese/base catalytic systems that facilitate a new mode of C–H bond activation by manganese via a redox-neutral base-assisted deprotonation mechanism. As such, the requirement for the tedious preparation of MnR(CO)5 complexes (R = Me, Bn, Ph) in stoichiometric reactions was eliminated, and a series of manganese-catalyzed C–H activation reactions of arenes with various reaction partners having C≡C and C═C bonds were achieved. Through the latter strategy of synergy between manganese catalysts (with or w/o bases) and acids, we disclosed a “dual activation” mode for performing manganese-catalyzed C–H bond transformations, that is, merging C–H activation by manganese catalysts and C–X multiple bond activation by Lewis acids. Consequently, the scope of C–H substrates could be expanded to include challenging ketones and olefinic C–H compounds. Additionally, the range of reaction partners could be significantly broadened to include those bearing more polarized C═O, C═N, and C≡N bonds such as aldehydes, imines, and nitriles. Remarkably, the innate reactivity of different C–H bonds in ketones could be reversed by manganese catalysis, and the reactions could even be carried out at room temperature. Our findings provide guiding information for the future development of manganese-catalyzed C–H activation reactions and beyond. Related important contributions from other groups are mentioned, and the remaining challenges and future perspective in this emerging area are also presented.
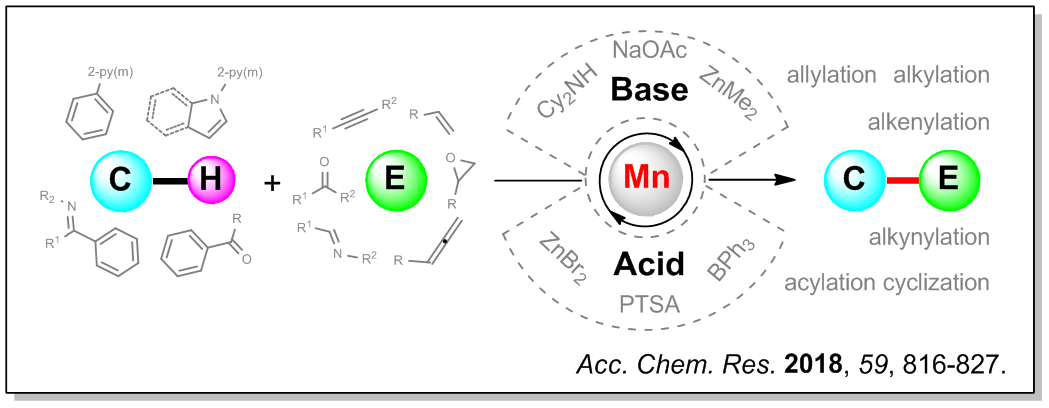
This work has been published in Acc. Chem. Res. 2018, 51, 816-827.